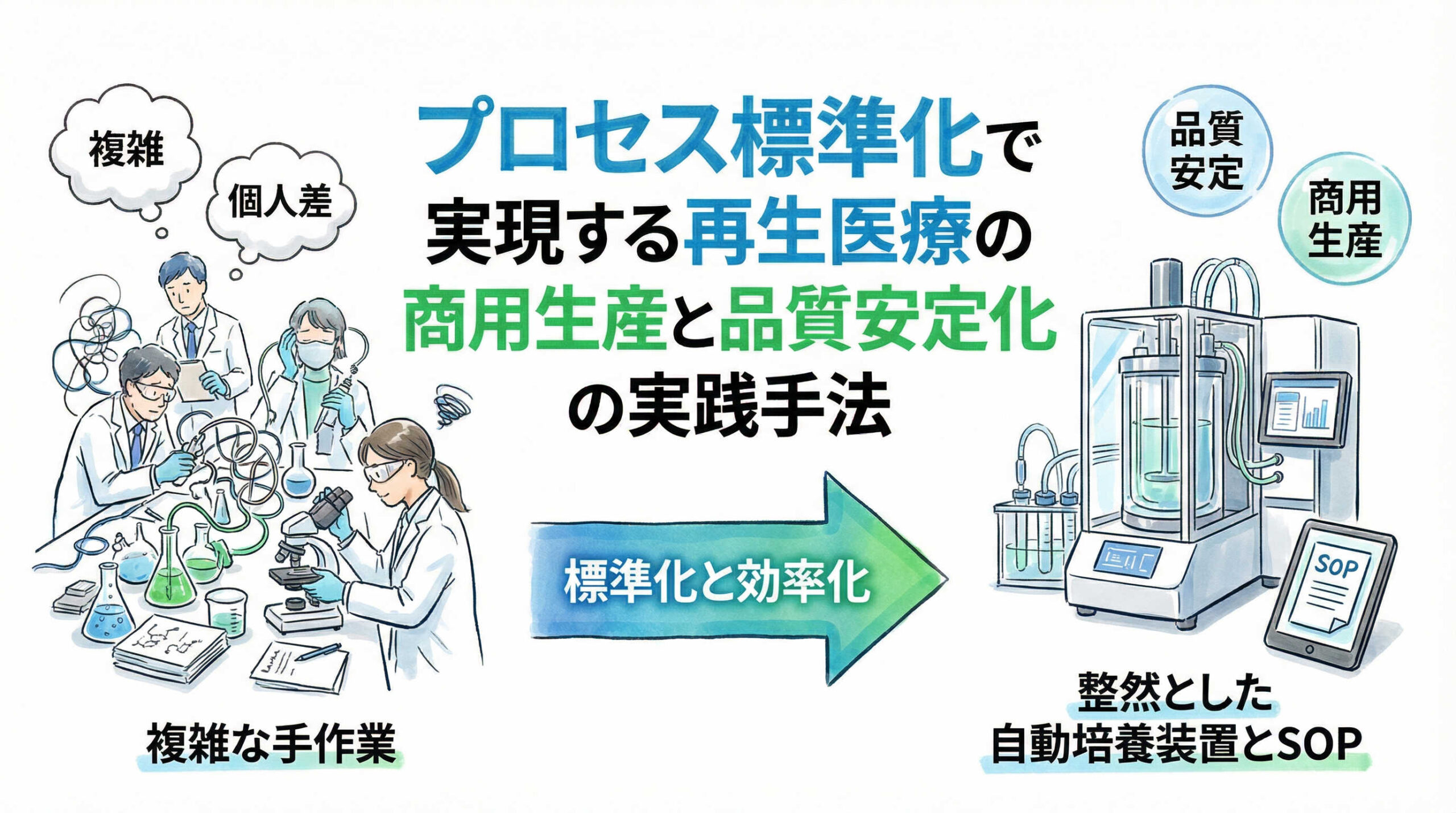
再生医療製品の研究開発が進み、いよいよラボスケールから臨床、そして商用生産へとステージを移す際、多くの現場担当者が直面するのが「製造プロセスの再現性」という壁ではないでしょうか。
研究室では熟練の研究員の手技によって高品質な細胞加工物が製造できていても、規模拡大や人員増加に伴い、品質のばらつきや属人化といった課題が浮き彫りになるケースは少なくありません。
再生医療における「プロセス標準化」は、単なる作業手順の統一にとどまらず、GCTP/GMPといった規制要件への適合や、将来的なコストダウン、技術移転の成功を左右する極めて重要な経営課題とも言えます。
本記事では、再生医療の商用化を見据えたプロセス標準化の定義から、QbD(Quality by Design)に基づくアプローチ、現場でのSOP作成のポイント、そして自動化技術の活用まで、実務に即した具体的な手法を解説します。
安定した品質の製品を患者様に届けるための基盤づくりとして、ぜひ参考にしてください。
再生医療におけるプロセス標準化とは?商用化に向けた定義と重要性

再生医療分野において「プロセス標準化」という言葉を耳にする機会は増えましたが、その本質的な定義と商用化における重要性を改めて整理してみましょう。
単にマニュアルを作成することだけが標準化ではありません。それは、いつ、誰が、どこで製造しても、科学的に同等な品質を持つ製品(細胞加工物)を安定的に供給できる状態を作り上げることを意味します。
ここでは、商用化を見据えた際に特に重要となる3つの観点から解説します。
製造工程の均一化による品質恒常性の確保
再生医療製品、特に生きた細胞を扱う場合、原材料である細胞そのものの個体差や状態変化が避けられません。そのため、製造工程(プロセス)自体を厳格にコントロールし、均一化することが極めて重要になります。
プロセス標準化によって、作業者の手技や判断によるブレを最小限に抑えることができれば、最終製品の品質恒常性(Consistency)を確保しやすくなります。これは、製品の安全性と有効性を担保するための最初の一歩と言えるでしょう。
GCTP/GMP省令に対応した製造管理・品質管理の基盤
商用化を目指す上で避けて通れないのが、GCTP(再生医療等製品の製造管理及び品質管理の基準に関する省令)やGMPへの適合です。これらの規制要件では、あらかじめ定められた手順通りに製造され、その記録が正確に残されていることが求められます。
プロセスが標準化されていなければ、バリデーション(妥当性確認)を実施することも、逸脱管理を適切に行うことも困難です。つまり、プロセス標準化は、規制当局に対して製品の品質を保証するための法的・制度的な基盤でもあるのです。
ラボスケールから商用生産へ移行する際の再現性担保
研究開発段階の小規模な製造(ラボスケール)では成功していても、製造規模を拡大したり、複数の拠点で製造したりするフェーズになると、途端に再現性が失われることがあります。
これは、ラボレベルでは「暗黙知」として処理されていた細かな条件や手技が、明文化されていないことに起因する場合が多いです。プロセス標準化は、ラボでの成功体験を商用生産という大規模な環境でも確実に再現するために不可欠な翻訳作業とも言えます。
なぜ今、再生医療の製造プロセス標準化が必要なのか

再生医療の産業化が加速する現在、なぜこれほどまでに製造プロセスの標準化が叫ばれているのでしょうか。
それは、従来の低分子医薬品やバイオ医薬品とは異なり、再生医療等製品特有の「不安定さ」や「複雑さ」が、ビジネスの成否に直結するからです。
ここでは、製造現場が抱える具体的な課題と、標準化によって得られるメリットを4つの側面から掘り下げていきます。
手作業による技術の属人化と品質ばらつきの排除
多くの培養現場では、依然として熟練技術者の「神の手」に依存している工程が存在します。しかし、特定の人物しか成功しないプロセスでは、商用生産は成り立ちません。
プロセス標準化の最大の目的の一つは、この属人化の排除です。手技のスピード、ピペッティングの角度、細胞の観察基準などを定量化・言語化することで、経験の浅い作業者でも熟練者と同じ結果が出せる体制を構築する必要があります。これにより、品質のばらつき(Variability)を大幅に低減できるでしょう。
製造コストの適正化と歩留まり(収率)の改善
再生医療製品の製造コストは非常に高く、その多くを人件費や培地・試薬代が占めています。プロセスが不安定で失敗(バッチの廃棄)が発生すれば、その損失は甚大です。
標準化されたプロセスは、ミスの発生率を下げ、歩留まり(収率)を向上させます。また、作業効率が上がることで、限られたリソースでより多くの製品を製造できるようになり、結果として製造原価の低減、ひいては薬価への反映や事業収益性の向上につながります。
技術移転(テックトランスファー)の円滑化
自社で開発したシーズをCDMO(医薬品製造受託機関)へ委託する場合や、海外展開のために現地製造を行う場合、「技術移転(テックトランスファー)」が発生します。
この際、プロセスが曖昧なままだと移転先で再現できず、開発スケジュールが大幅に遅延するリスクがあります。標準化され、文書化された堅牢なプロセスパッケージがあれば、技術移転はスムーズに進み、市場投入までの時間を短縮することが可能になります。
変更管理およびバリデーション負荷の軽減
製造販売承認後であっても、原材料の変更や設備の更新など、製造プロセスの変更が必要になることは多々あります。
このとき、元のプロセスが標準化され、管理された状態(State of Control)にあれば、変更による品質への影響評価や、必要なバリデーションの範囲を論理的に決定することができます。逆に標準がない状態での変更は、何が影響したのか特定できず、膨大な再検証コストを招くことになりかねません。
プロセス標準化を実現するQbD(Quality by Design)アプローチ

プロセス標準化を科学的かつ合理的に進めるための手法として、医薬品開発で推奨されているのが「QbD(Quality by Design:品質・バイ・デザイン)」アプローチです。
これは「品質は試験によってではなく、設計によって作り込まれるべきである」という考え方に基づいています。
経験と勘に頼るのではなく、データとリスク評価に基づいてプロセスを設計するQbDの流れを確認していきましょう。
TPP(目標製品プロファイル)に基づく目標品質の設定
QbDの出発点は、「どのような製品を作りたいか」というゴールを明確にすることです。これをTPP(目標製品プロファイル)やQTPP(目標製品品質プロファイル)と呼びます。
具体的には、対象疾患、投与経路、用量、安全性プロファイルなどを定義します。このゴール設定が曖昧だと、どの工程をどこまで厳密に管理すべきかの基準が定まらず、効果的な標準化が行えません。まずは「あるべき姿」を定義することから始めましょう。
CQA(重要品質特性)の特定とリスク評価
次に、設定した目標品質を達成するために不可欠な製品の特性を特定します。これをCQA(重要品質特性)と呼び、細胞数、生存率、純度、力価(Potency)、無菌性などが該当します。
そして、各CQAに対して、どの工程がどの程度影響を与えるかをリスクアセスメントによって評価します。すべての特性を同じ強度で管理するのではなく、リスクの高い項目にリソースを集中させることで、効率的な管理が可能になります。
CPP(重要工程パラメータ)の抽出とデザインスペースの構築
CQAに影響を与える製造パラメータ(温度、pH、培養時間、試薬濃度など)の中から、特に管理が必要なものをCPP(重要工程パラメータ)として抽出します。
単に一点の最適値を決めるだけでなく、品質が許容範囲内に収まるパラメータの組み合わせ範囲(デザインスペース)を実験計画法(DoE)などを用いて構築します。この「幅」を持った設計こそが、商用生産におけるプロセスの柔軟性と堅牢性を両立させる鍵となります。
管理戦略の策定と継続的なプロセス確認
CPPとCQAの関係性が理解できたら、それらを日常的にどう管理するかという「管理戦略」を策定します。これには、工程内管理試験(IPC)の設定や、稼働性能適格性確認(PQ)などが含まれます。
さらに、商用生産開始後も継続的なプロセス確認(CPV)を行い、蓄積されたデータをモニタリングすることで、プロセスが管理状態にあることを常に保証し続ける姿勢が求められます。
現場レベルでの標準化推進ステップとSOP作成のポイント

QbDで設計されたプロセスを、実際の製造現場(GMP施設など)で運用可能な形に落とし込むには、SOP(標準作業手順書)の整備と教育訓練が欠かせません。
しかし、現場では「SOP通りにやったはずなのにうまくいかない」という声がよく聞かれます。
ここでは、実効性のあるSOP作成と、現場への定着を図るための具体的なステップについて解説します。
製造フローの細分化と熟練者による暗黙知の形式知化
まず行うべきは、現在の製造フローを最小単位の操作(ユニットオペレーション)まで分解することです。そして、熟練者が無意識に行っているコツや判断基準(暗黙知)を洗い出します。
例えば、「細胞が元気な状態」とは具体的にどのような形態か、「優しくピペッティング」とは何秒で何ml吸うことか。これらをヒアリングや観察によって徹底的に形式知化することが、標準化の第一歩です。
定量的な判断基準を設けたSOP(標準作業手順書)の整備
形式知化された情報を基にSOPを作成しますが、ここで重要なのは曖昧な表現を排除することです。「適当に」「十分に」「速やかに」といった言葉は、作業者によって解釈が異なります。
「5分以内に」「3回反転混和する」「色が〇〇になるまで」といった、誰が読んでも同じ行動をとれる定量的な記述を心がけましょう。判断に迷う箇所をなくすことが、ヒューマンエラー防止に直結します。
画像や動画を活用した教育訓練と力量評価の仕組み
文字だけのSOPでは、ニュアンスが伝わりにくい手技も多々あります。特に細胞培養のような繊細な作業では、動画や写真を用いたビジュアルマニュアルが非常に有効です。
また、SOPを読ませるだけでなく、実際に手技を行わせ、合格基準を満たした者だけを作業認定する「力量評価」の仕組みを導入しましょう。定期的な再評価を行うことで、自己流への回帰(ドリフト)を防ぐことができます。
原材料・資材の規格統一とサプライヤー管理の徹底
プロセス標準化において見落とされがちなのが、インプット(原材料)の管理です。作業手順が完璧でも、使用する培地ロットやプラスチック消耗品の仕様が微妙に異なれば、結果は変わってしまいます。
重要原材料については複数のサプライヤーを確保しつつも、規格を厳格に統一し、サプライヤー変更時の評価手順(チェンジコントロール)を確立しておくことが、安定生産の要となります。
自動培養装置・閉鎖系システムの導入による標準化の高度化

手作業の標準化には限界があります。そこで近年、プロセス標準化の強力なソリューションとして導入が進んでいるのが、自動培養装置や閉鎖系システムです。
これらは単に省力化のためだけでなく、品質の再現性を極限まで高めるための手段として注目されています。
ハードウェアの導入がもたらす標準化の高度化について、具体的なメリットを見ていきましょう。
手技の介在を最小化することによる人為的ミスの防止
自動化の最大のメリットは、機械がプログラム通りに動作するため、疲労や集中力低下によるヒューマンエラーが排除されることです。
ピペッティングの速度、撹拌の強さ、タイミングなどが常に一定に保たれるため、手作業では達成困難なレベルでの再現性が実現します。特に、長期間にわたる培養工程や、複雑な操作を要する工程において、その効果は絶大です。
培養環境の厳密な制御とリアルタイムモニタリング
最新の自動培養装置には、pH、溶存酸素(DO)、温度、グルコース濃度などをリアルタイムで測定・制御するセンサーが搭載されています。
インキュベーターの開閉による環境変化のリスクをなくし、細胞にとって常に最適な環境を維持し続けることができます。これにより、細胞の状態変化を最小限に抑え、ロット間の品質ばらつきを大幅に低減することが可能です。
データインテグリティの確保と製造記録の自動化
GMP環境下では、データインテグリティ(DI:データの完全性)が厳しく求められます。手書きの記録では記入ミスや改ざんのリスクが残りますが、自動装置であれば製造パラメータや操作ログが自動的に電子記録として保存されます。
これは監査対応の負担を軽減するだけでなく、万が一トラブルが発生した際の原因究明(トラブルシューティング)においても、強力なトレーサビリティを提供します。
コンタミネーションリスクの低減と無菌操作の確実性
開放系での操作は、常にコンタミネーション(汚染)のリスクと隣り合わせです。閉鎖系システム(Closed System)や自動装置を導入することで、細胞が外気に触れる機会を遮断できます。
これにより、グレードの高いクリーンルーム(グレードA/B)での作業時間を減らし、設備維持コストを下げつつ、無菌操作の確実性を担保することが可能になります。これは製品の安全性確保において非常に大きなアドバンテージです。
プロセス標準化において直面しやすい課題と対策

ここまでプロセス標準化の重要性と手法を解説してきましたが、実際の運用では一筋縄ではいかない課題にも直面します。
再生医療特有の難しさを理解し、あらかじめ対策を講じておくことが、プロジェクトの停滞を防ぐ鍵となります。
ここでは、現場で特によくある3つの課題と、その向き合い方について触れておきます。
細胞ソース(ドナー)ごとの個体差への対応策
自家細胞を用いた治療などでは、ドナーの年齢や健康状態によって細胞の増殖能や特性が大きく異なります。プロセスを完全に固定してしまうと、増殖が遅い細胞に対応できず、規格外品となってしまう恐れがあります。
対策としては、培養期間や播種密度にある程度の幅(許容範囲)を持たせたデザインスペースを設定するか、あるいは細胞の状態に応じてフィードバック制御を行う「適応型製造(Adaptive Manufacturing)」の概念を取り入れることが検討されます。
プロセスの柔軟性と厳格な管理基準のバランス
標準化と聞くと「一切の変更を許さない厳格さ」をイメージしがちですが、研究開発段階ではプロセスの改善や最適化も必要です。
初期段階では変更の余地を残した柔軟な管理を行い、臨床試験が進むにつれて段階的に管理基準を厳格化していく「段階的な標準化」のアプローチが有効です。開発フェーズに応じた適切な管理レベルを見極めるバランス感覚が求められます。
製造変更時における同等性/同質性評価のハードル
製造コスト削減やスケールアップのために、培養容器や培地を変更したい場面は必ず訪れます。しかし、標準化されたプロセスの変更には、変更前後で製品品質が変わっていないことを証明する「同等性/同質性評価」が必要です。
このハードルを見越し、開発初期から将来のスケールアップを想定した機器選定を行ったり、比較可能性(Comparability)を評価するためのバックアップサンプルを確保しておいたりする戦略的な準備が不可欠です。
まとめ

再生医療におけるプロセス標準化は、高品質な製品を安定的に患者様へ届けるための必須条件であり、商用化成功の鍵を握る重要な取り組みです。
手作業の属人化を排除し、QbDアプローチに基づく科学的なプロセス設計を行うこと、そして現場レベルでのSOP整備や自動化技術の導入を進めることで、製造の再現性は飛躍的に向上します。
重要なポイントを振り返りましょう。
- 定義: プロセス標準化は、品質恒常性の確保と規制対応の基盤である。
- 目的: 属人化の排除、コスト適正化、技術移転の円滑化を目指す。
- 手法: QbDによる設計、定量的なSOP、自動化システムの活用が有効。
- 課題: 原材料のばらつきや変更管理の難しさを理解し、柔軟性と厳格さのバランスを取る。
標準化は一度完成させれば終わりではありません。製造データを継続的にモニタリングし、プロセスを見直し続ける終わりのない改善活動です。
この記事が、貴社の製造プロセスをより強固なものにし、再生医療の発展に寄与する一助となれば幸いです。
プロセス標準化についてよくある質問

以下に、再生医療のプロセス標準化に関してよく寄せられる質問とその回答をまとめました。実務の参考にしてください。
- Q1. プロセス標準化はいつから始めるべきですか?
- 早ければ早いほど良いですが、本格的には非臨床試験(GLP試験)に使用する検体を製造する段階から意識すべきです。臨床試験(治験)入りする段階では、GCTPに準拠した標準化が必須となります。
- Q2. 手作業のままでも標準化は可能ですか?
- 可能です。ただし、作業者の技量に依存するリスクが残ります。徹底した教育訓練、詳細なSOP、ダブルチェック体制などでカバーする必要がありますが、将来的には自動化も検討することをお勧めします。
- Q3. QbDアプローチは必須ですか?
- 規制上、厳密に「QbDでなければならない」とは明記されていませんが、現在の承認審査のトレンドや、変更管理の合理性を説明するためには、事実上の標準(デファクトスタンダード)となっています。
- Q4. 自動化装置導入のコストメリットは?
- 初期投資は高額ですが、人件費の削減、ヒューマンエラーによる廃棄ロスの低減、クリーンルームの維持費削減などを長期的に見れば、十分なコストメリットが出るケースが多いです。
- Q5. 既存のプロセスを変更する場合の注意点は?
- 変更前後の製品の品質が同等であることを証明する必要があります。リスクアセスメントを行い、必要な試験項目を設定して、データの比較評価(同等性評価)を慎重に行ってください。